-
23 November 2025 | Country Update
A new procedure for faster access to innovative pharmaceuticals -
27 May 2025 | Country Update
Implications of patient representatives for the reimbursement of medical products -
24 January 2025 | Country Update
Reimbursement for import or replacement costs of unavailable essential medicines -
02 September 2024 | Country Update
Modernizing pharmaceutical reimbursement procedures to ensure rapid and sustainable access to medicines -
08 March 2024 | Country Update
A new purchasing policy for biosimilars in hospitals: A mandatory tendering process -
04 August 2023 | Country Update
Medical mobile applications: A new procedure for reimbursement -
20 January 2023 | Country Update
Royal decree allowing for limits on exports of medicines in case of crisis
2.4. Regulation and planning
2.4.1. Regulation and governance of third-party payers
Compulsory health insurance is administered by sickness funds, which are private non-profit-making organizations with a public interest mission controlled by the Supervising Authority for Sickness Funds and National Associations of Sickness Funds (see Fig2.1) (Sickness Funds Act; BS-MB 28 September 1990).
Fig 2.1
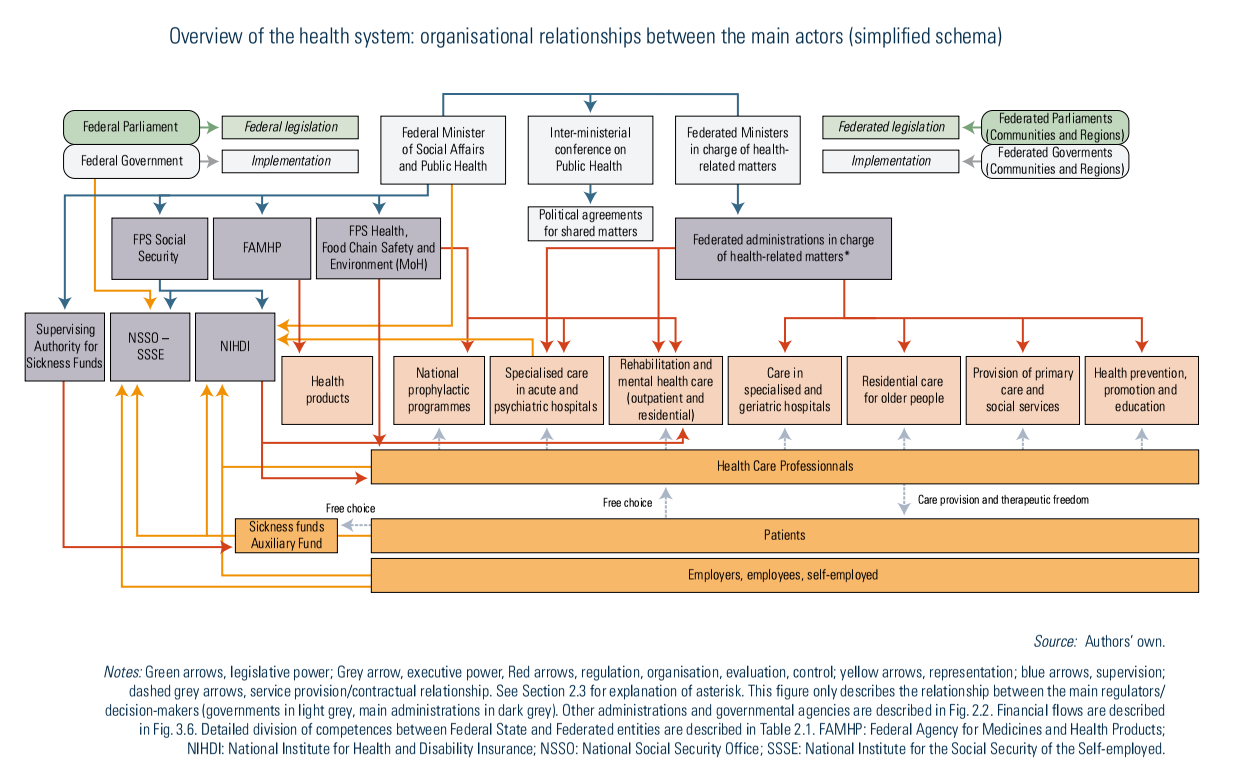
All individuals entitled to health insurance must register with a sickness fund. The choice is free, except for railway workers, who are automatically covered by the health insurance fund of the Belgian railway company. Sickness funds are mainly organized according to religious or political affiliations into five national alliances: the National Alliance of Christian Mutualities, the National Union of Neutral Mutualities, the National Union of Socialist Mutualities, the National Union of Liberal Mutualities and the National Union of the Free Mutualities (NIHDI, 2014b). In 2019, the National Alliance of Christian Mutualities and the National Union of Socialist Mutualities together had the largest share of the general system, covering about 40.7% and 28.5% of the population, respectively.
Besides the management of the compulsory health insurance, they provide a mandatory entitlement to complementary advantages on services like orthodontics or homeopathy and VHI, for example to cover for extra-billing for a single room in hospital (see Section 3.5). Members have to pay an additional flat-rate contribution (a community-rated premium) for these complementary advantages and services. Otherwise, people who do not want these mandatory complementary services must enrol in the Auxiliary Fund, an additional neutral public body that only manages the compulsory health insurance (0.95% of the population). People who have not registered with a sickness fund will be affiliated to this Auxiliary Fund.
Competition among sickness funds concentrates mainly on their mandatory complementary services. Legally, sickness fund members have the opportunity to change their sickness fund each quarter if they have been enrolled for a period of at least one year, but insurance mobility in practice remains limited (Schokkaert et al., 2003).
Sickness funds receive a prospective budget to finance the health care costs of their members and their accountability has been increased over time (see Box3.5 in Chapter 3).
2.4.2. Regulation and governance of provision
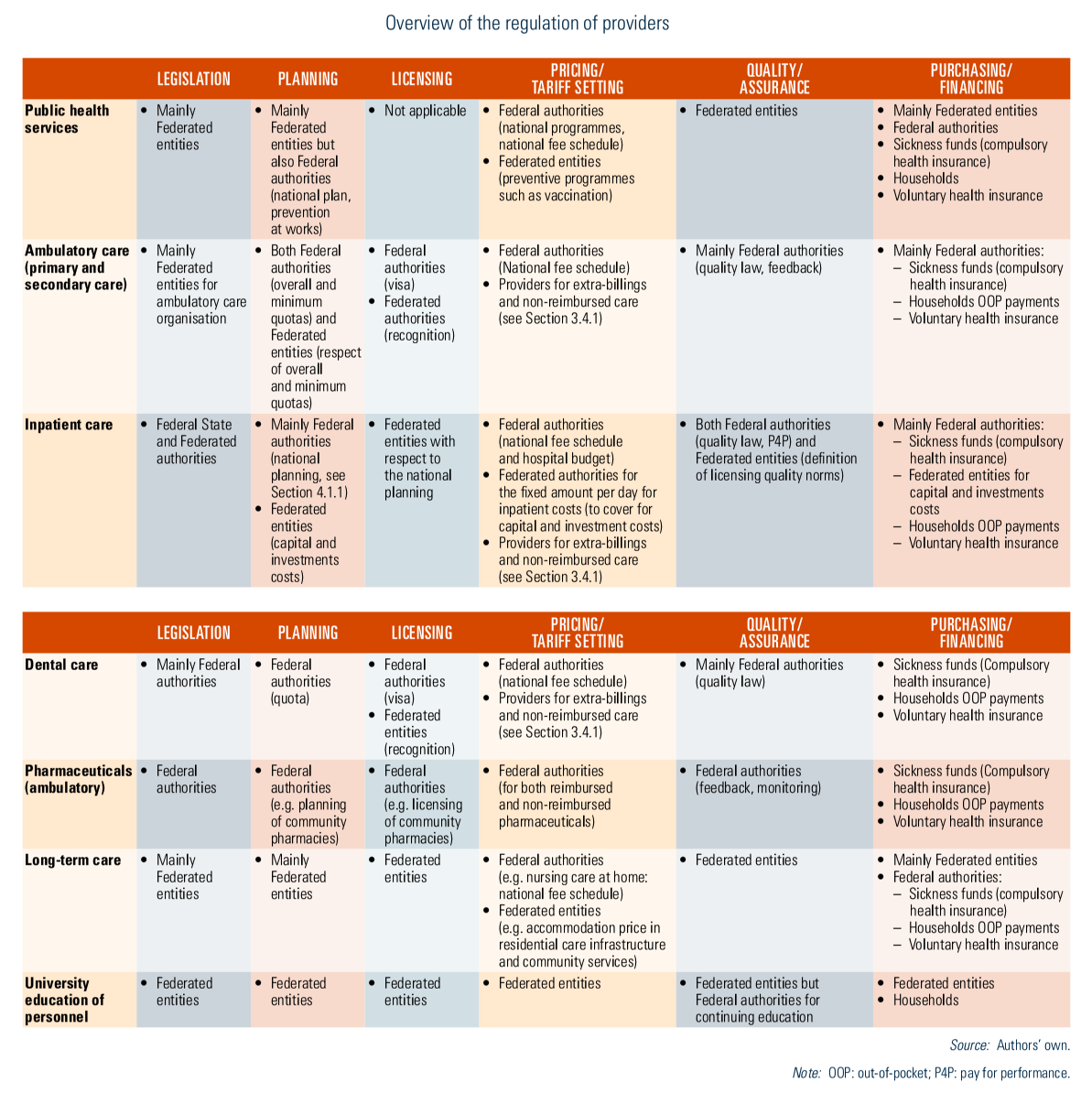
Reimbursed health care services are provided by both public and private facilities, and individual health professionals who mainly comply with the same set of rules, enjoy the same therapeutic freedom and offer the same services. For patients, the difference between the two sectors lies mainly in the extra-billings charged. An overview of the regulation of providers is described in Table2.4.
Table 2.4
Health facilities
For hospitals, national planning has been established at the Federal level, defining programming criteria such as the number of beds per 100 000 inhabitants or, in the case of maternity wards, per 1000 births and based on the size, age pyramid and morbidity of the target population, as well as on geographical distribution. In 1982, the government decided to freeze the number of licensed beds for all general hospitals (BS-MB 7 November 1964). This rule is still in effect today, with the creation of a new bed necessarily being accompanied by the closure of another. Programming criteria have also been introduced for heavy medical equipment, some medical and medico-technical services and some care programmes (MoH, 2016c). More recently, the project of reform of the hospital payment system, launched in April 2015, includes a reflection on determining capacity planning according to the needs of the population and scientific evidence (Van de Voorde et al., 2017).
Besides programming criteria, hospitals, services, functions and care programmes must also comply with licensing criteria that ensure quality of care (Justel 7 November 2008). Each hospital, service and care programme must be licensed by the Federated authorities to receive financing (from both Federal State and Federated entities). Federal authorities have defined basic organizational rules and Federated entities are competent to define licensing rules in accordance with the national planning and the Federal basic organizational rules. The Federal State may exercise its veto if these rules have a negative impact on the Federal budget. Licensing criteria are of different types: organizational norms related to staff requirements (for example, qualification levels, ratio between qualified personnel and auxiliaries) and responsibilities (for example, hygiene, ethics); architectural criteria (for example, the number, size and hygiene standard of rooms); functional standards (for example, convenience, accessibility); minimum activity and facility standards; and expected staff numbers (BS-MB 7 November 1964).
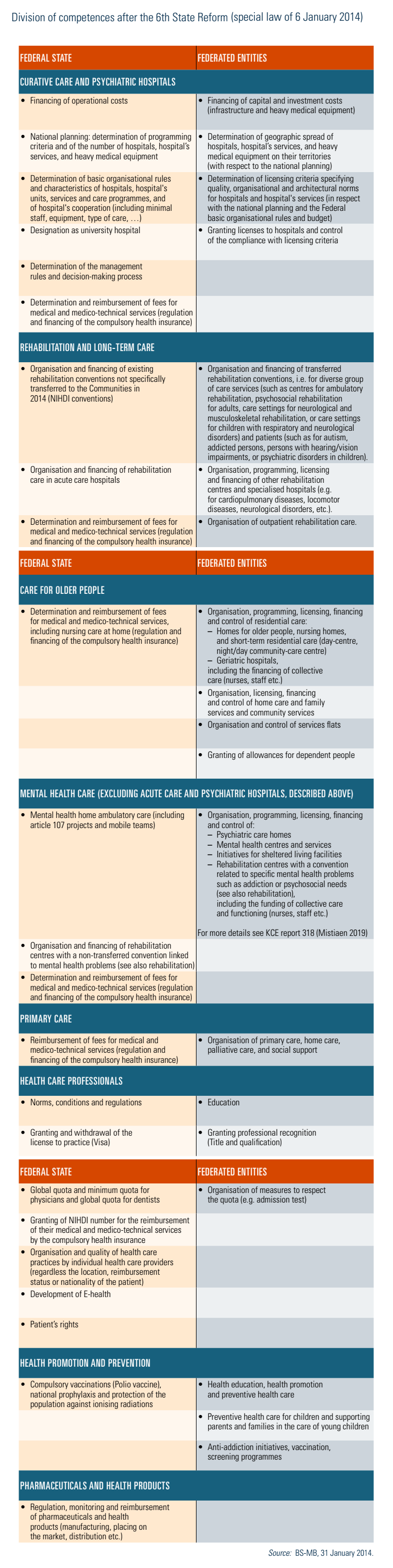
Regulation of other health facilities, such as homes for older people, nursing homes or mental care centres, is the responsibility of Federated entities (see Table2.1).
Table2.1
In addition, health care facilities can apply voluntarily for accreditation, for an external evaluation (for example, Canada international accreditation) (see Section 5.4.2). Some other quality labels can also be requested voluntarily, such as Baby-Friendly (MoH, 2016d) for hospitals.
Additional quality initiatives are described in Box2.4.
Box 2.4

Health care professionals
The practice of health care professionals, such as physicians, dentists, physiotherapists, pharmacists, nurses, midwives and other practitioners of a paramedical profession, is regulated by the Practice of Health Care Professions Act (BS-MB 18 June 2015) (see Section 4.2.1). The planning of health care professionals is described in the next subsection. The organization of education is a competence for the Flemish and French communities. Some training aspects differ between the two communities (see Section 4.2.4). Quality initiatives are described in Box2.5.
Planning
Evaluation of needs concerning the supply of human resources for health is a Federal competence. The Planning Commission of Medical Supply, created in 1996, is responsible for assessing medical workforce needs in the professions of physicians, dentists, physiotherapists, nurses, midwives and speech therapists (BS-MB 29 August 1996). The Commission provides non-binding opinions on human resource planning to the Minister of Social Affairs and Public Health. The Planning Unit for the Supply of Health Care Professions brings administrative and technical support to the Planning Commission. The Planning Unit quantifies and predicts the staffing and workforce needs of health care professionals using modelling (see Box2.3) (Benahmed et al., 2019).
Based on the advice of the Planning Commission, a system of quotas for physicians and dentists is established. An overall quota is set up for Belgium and stratified by community (sub-quotas). Sub-quotas are managed by the Federated entities, which are currently creating their own planning commissions to manage them (see Box4.3 in Chapter 4) (BS-MB 31 January 2014). There is also national planning for inpatient care, which determines for instance the maximum number of beds per hospital service and the maximum amount of heavy medical equipment (see Section 2.4.2).
2.4.3. Regulation of services and goods
Basic benefit package
The services that are (partially) covered by compulsory health insurance are described in the national fee schedule (called the nomenclature) and can be found on the NIHDI website (https://www.riziv.fgov.be/fr/nomenclature/Pages/default.aspx). The fee schedule is negotiated yearly or biennially between representatives of the sickness funds and of health care professionals (see Section 3.3).
In 2019, the NIHDI began a major project: the structural reform of the fee schedule for physicians (see Section 6.1).
Health technology assessment
Since 2002, the KCE was established with health technology assessment as one of its core activities and is recognized as the national reference in this field (KCE, 2019).
The KCE imposes rigorous scientific procedures based on international standards and using external expert reviewers (so-called validators), while paying attention to potential conflicts of interest. Each report is submitted for approval to the board of directors, which is composed of representatives of the different health care stakeholders and includes recommendations for health policy-makers. The KCE is involved in neither the policy decisions nor their implementation. The KCE is for instance not implicated in the reimbursement decision process for pharmaceuticals but can perform a health technology assessment on a pharmaceutical at the NIHDI’s request.
All reports are published and can be downloaded free of charge. For the 2010–2019 period, 200 reports have been published, including 45 health technology assessments.
Regulation of medical devices and aids
Before placing a device on the market, manufacturers must undertake a conformity assessment of the device, according to a procedure related to their risk class and affix the CE marking. For higher-risk devices, the intervention of a notified body is necessary to perform the conformity assessment and obtain a certificate. Notified bodies are designated and supervised by the FAMHP. In addition to the mandatory CE marking, a registration of the distributors and exporters including limited information on their products to the FAMHP is necessary for traceability reasons (BS-MB 20 December 2013; BS-MB 7 December 2017; BS-MB 26 April 2019).
Every incident with a medical device must be notified to the FAMHP. Manufacturers must have a vigilance system and report the incidents. Belgian hospitals and distributors including community pharmacies must have a contact point for performing this notification (BS-MB 7 December 2017). The FAMHP evaluates these incidents and the proposed corrective and preventive actions to prevent reoccurrence.
Concerning distribution, liberalization began in 2019 (BS-MB 28 January 2019) and some risk-free products such as sterile dressings, probes, heat ointments and cooling sprays can now be distributed in supermarkets and are no longer restricted to community and hospital pharmacies.
In the past, some criticism had been expressed regarding the regulation of medical devices, which was considered as less stringent than for pharmaceuticals because pharmaceuticals must prove efficacy while medical devices only have to prove safety and performance (Hulstaert et al., 2012). In 2017, new regulations were issued ((EU) 2017/745 for medical devices and (EU) 2017/746 for in vitro diagnostic medical devices) and tighter controls were imposed on high-risk devices, including registration in the Eudamed system, the use of a unique code for traceability and the increase of communication between Member States. Controls will also be tightened on clinical trials and on the notified bodies (European Council, 2017). All vigilance reports, including incidents, need to be reported in Eudamed.
Placing a device on the market does not automatically imply reimbursement from the compulsory health insurance. For the reimbursement process, a distinction is made between invasive and non-invasive devices and the decision is made in different committees and commissions. An optimization of the procedure is foreseen for 2020, for example with the creation of a single committee for different domains.
For invasive devices and implants, the added value must be proven with scientific studies, and health economic and epidemiological data. Reimbursement criteria include the importance of the device in medical practice based on therapeutic and social needs, the budgetary impact, and the ratio between the costs for insurance and the therapeutic value of the device (BS-MB 1 July 2014). The defined reimbursement modalities include:
- the reimbursement basis (which may differ from the public price);
- the reimbursement category (invasive device or implant) and two additional sub-categories defining the method of reimbursement (such as a lump sum or not, included in a nominative list or not) and patient contributions (see Section 3.4.1);
- the reimbursement conditions; and
- the security margin, for example, a percentage of the reimbursement basis defining the maximum authorized public price to be reimbursed (NIHDI, 2019q; Vinck et al., 2018).
For mobility aids such as wheelchairs, reimbursement is transferred from the Federal level to the Federated entities due to the 6th State Reform. The particular regulations regarding mobile health applications can be found at https://mhealthbelgium.be/.
Authors
References
2.4.4. Regulation and governance of pharmaceuticals
This section focuses on pharmaceuticals for human use, which are mainly regulated by the Medicines Act (BS-MB 17 April 1964). The FAMHP is the main institution for the regulation of pharmaceuticals and is responsible for the quality, safety and efficacy of pharmaceuticals and health products. FAMHP is responsible (FAMHP, 2019a) for the assessment (including authorization) and monitoring of:
- clinical trial requests;
- registration and market authorization (MA) requests;
- manufacture, distribution, delivery, imports and exports;
- pharmacists’ activities;
- pharmacovigilance;
- proper patient information and health care professional information on health products; and
- advertising activities.
Market authorization
Market authorization (MA) is under the responsibility of the Minister of Social Affairs and Public Health, and is based on the recommendations of FAMHP. The first MA is valid for five years, after which renewal can be requested. After renewal, the MA is valid indefinitely, except when there is still doubt on safety or by application of the Sunset Clause (FAMHP, 2019c). During the MA or registration procedure, pharmaceuticals are classified into prescribing and non-prescribing pharmaceuticals (over-the-counter pharmaceuticals) (BS-MB 17 April 1964).
Quality and pharmacovigilance
Every MA holder needs a qualified person to control the quality of produced and imported products. They also need a qualified person responsible for establishing and operating the pharmacovigilance system ( BS-MB 22 December 2006).
They are obliged to record all suspected adverse reactions in the European database EudraVigilance (EMA, 2018).
Also, health care professionals and patients are encouraged to report any adverse reactions they observe to the FAMHP web portal (www.eenbijwerkingmelden.be (article 67bis; BS-MB 22 December 2006), before the FAMHP transmits them to the EudraVigilance database.
Communication on pharmacovigilance is available on the FAMHP website (VIG-News, Direct Healthcare Professional Communications). In addition, dedicated communication for health care professionals is carried out in collaboration with the Belgian Centre for Pharmacotherapeutic Information, via a website (www.bcfi.be) and a smartphone application.
Distribution
In contrast to some other countries, pharmaceuticals are not available via supermarkets in Belgium. The law provides a monopoly for distribution of pharmaceuticals (prescription and over-the-counter) via community and hospital pharmacies. Some exceptions are allowed for physicians to deliver pharmaceuticals, such as in urgent cases, for samples of pharmaceuticals, for compassionate use, for clinical trials and for prophylaxis campaigns against infectious diseases (BS-MB 14 November 1967; BS-MB 18 June 2015). Distribution via the Internet is possible for registered over-the-counter pharmaceuticals, but only when a Belgian community pharmacy owns the website (BS-MB 30 January 2009).
Wholesaling of pharmaceuticals is submitted to licence issued by the Minister of Social Affairs and Public Health via the FAMHP (BS-MB 22 December 2006). Wholesalers with a public service obligation, called full-line wholesalers (Grossistes-répartiteurs/Groothandelaars-verdelers), are the main suppliers of the daily distribution of all authorized medicines in Belgium and have the legal obligation to deliver products within 24 hours to the pharmacist (BS-MB 22 December 2006). Pharmaceutical companies have the obligation to deliver within three working days to the full-line wholesalers and pharmacists (BS-MB 03 February 2020).
The opening, transfer or merger of community pharmacies must be licensed by the Minister of Social Affairs and Public Health, and is submitted to demographic and geographical criteria to ensure an adequate supply (BS-MB 5 October 1974). To avoid oversupply, a moratorium on the number of community pharmacies was introduced in 1999, and is valid until 2023 (BS-MB 6 December 2019). Nevertheless, compared with other neighbouring countries Belgium has a high density of community pharmacies – 43.4 per 100 000 inhabitants in 2017 compared with an EU15 average of 28.8 (OECD, 2019c).
Measures to tackle pharmaceutical shortage are described in Box2.6.
Box 2.6

Advertising
Advertising on pharmaceuticals (including granting of advantages to health professionals) is subject to specific legislation. Any advertising to the general public on prescribing pharmaceuticals, narcotics and psychotropic is prohibited; see FAMHP (2019b) for more details.
Price setting
Price setting is under the responsibility of the Minister of Economic Affairs, based on the recommendations by the Committee of Pricing for Pharmaceutical Specialties of the FPS Economy (FPS Economy, 2019) (see Box2.7).
Box 2.7

The Commission for the Reimbursement of Medicinal Products (CRP) evaluates the reimbursement request based on five evaluation criteria: the therapeutic added value, the price and the proposed reimbursement basis, the importance of the medicine in medical practice in function of the therapeutic and social needs, the budgetary impact for the health insurance, and the ratio between the costs for the insurance and therapeutic value. A cost-effectiveness evaluation is only required when an added therapeutic value is claimed. There are also specific procedures for “orphan” drugs, biosimilar drugs and parallel imported drugs.
Box2.8 shows the composition of the public price of pharmaceuticals.
Box 2.8

Reimbursement
For prescribed pharmaceuticals, the applicant can request to be included in the positive list for reimbursed medicines. The procedure is examined at the same time as the price setting (see Fig2.4) by the CRP of the NIHDI, which formulates proposals to the Minister of Social Affairs and Public Health. Cost-sharing mechanisms for reimbursed pharmaceuticals are described in Section 3.4.1.
The CRP must give a proposal on the reimbursement to the Minister within 150 days and, based on this proposal, the Minister takes the final decision before day 180. If the pharmaceutical company does not receive a decision within 180 days, its application for reimbursement is automatically accepted (see Fig2.4).
Fig2.4
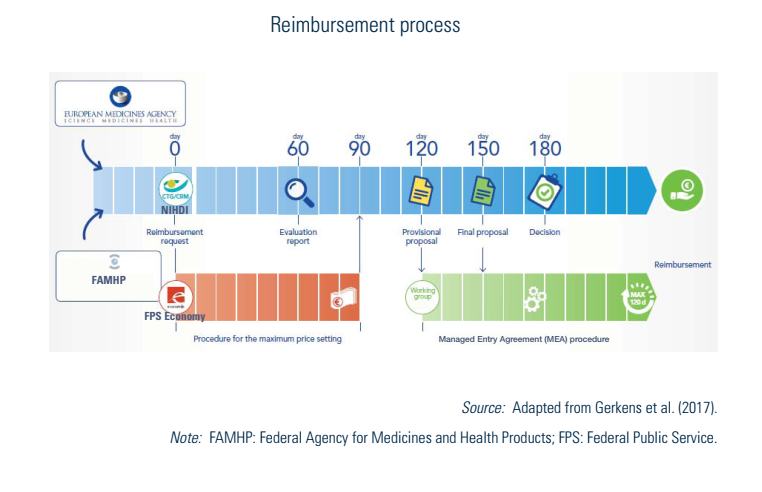
In case of uncertainty (clinical or economic) and for some legally defined pharmaceuticals (for example, orphan drugs or pharmaceuticals with a claimed added therapeutic value), managed entry agreements (MEAs) can be concluded at the applicant request, (i) when a reimbursement decision by the CRP was not possible at day 150; (ii) if proposed by the CRP during the process; or (iii) in case of negative decision of the CRP, after a motivated proposal of the Minister (see chapter V of the Royal Decree of 1 February 2018, BS-MB (15 March 2018)). Most of the time, they include financial compensation mechanisms that are confidential. MEAs have been possible since 2010 and conditions to conclude an MEA have been enlarged over time. Consequently, the procedures for conventions are rising and have become the rule rather than the exception for a majority of new innovative and expensive pharmaceuticals. The gross expenditure of pharmaceuticals under MEAs rose from €487 018 000 in 2014 (13% of total expenditure) to €1 430 953 000 in 2018 (31% of total expenditure). It should nevertheless be noted that actual expenditure for these MEAs are lower because of the agreed refunds. More details on MEAs can be found in the KCE Report 288 (Gerkens et al., 2017).
Authors
References
Since 1 April 2025, patient representatives have joined the Commission for the Reimbursement of Medical Products (CRP) to help better address the real needs of patients. The CRP now includes two representatives from patient associations – one from LUSS and one from VPP. While they are allowed to participate in discussions, they do not have voting rights. In addition, an external expert representing patients will be consulted during the evaluation of innovative medicines.
To further support this initiative, the NIHDI, LUSS and VPP have jointly developed a “patient questionnaire.” This tool is designed to gather insights into how the disease affects patients’ daily quality of life, as well as their experiences with current treatments and new medications.
Authors
Belgium is currently working on a reform of its pharmaceutical policy in consultation with industry and various stakeholders. The main objectives of the reform are to make the reimbursement process more demand-driven, to provide faster access to innovations with (potential) added value, to improve the assessment of this added value and of the evidence, to increase the transparency of the procedures, to involve patients more closely, and to make the procedures simpler and more efficient.
This reform includes the inclusion of international horizon-scanning initiatives, information on unmet medical needs and patient preferences, and real-world data in health technology assessment (HTA). One aspect of this reform also addresses confidential managed entry agreements for innovative medicines, with the aim of limiting their number, increasing their transparency, shortening their duration, and allowing companies developing generics and biosimilars to obtain confidential post-agreement price information in a timely manner.
Authors
References
To promote the use of biosimilars, the Royal Decree of 13 September 2023 introduced an obligation for hospitals to initiate a tendering process for the purchase of biologicals within nine months after the inclusion of an equivalent biosimilar (that is, a biosimilar with the same active ingredients at ATC5 level and the same method of administration) in the list of reimbursed medicines. The tendering period must be limited to two years and may only be renewed twice for 12 months if no new biosimilars are reimbursed. This Royal Decree also outlines a list of criteria that cannot be considered for the attribution of the procurement tender, such as the contractual link with other medicines.
Hospitals receive notification from NIHDI when a biosimilar is reimbursed, for example, on 4 March 2024, they were notified of the arrival of Ximluci (for eye treatment).
The NIHDI is also responsible for assessing this new procedure with a report conducted every two years. Discounts obtained as part of the public tendering procedure, however, will not be included in the assessment.
Authors
References
- Unavailability of the medicinal product has been either notified to the Belgian Federal Agency for Medicines and Health Products (FAMHP) or established by FAMHP;
- Unavailability is likely or certain for a minimum period of one month;
- Administration of the medicinal product is urgent and necessary, it has a major impact on the patient;
- Unavailability cannot (or cannot sufficiently) be substituted by other authorised medicines with the same therapeutic effect, regardless of the active substance.
For more details see: https://www.ejustice.just.fgov.be/eli/arrete/2023/01/19/2023030395/moniteur (French) / http://www.ejustice.just.fgov.be/eli/besluit/2023/01/19/2023030395/staatsblad (Dutch)